
- Publication: Nature
- Publication Date: August 26, 2021
- Organizations mentioned: DeepMind, Protein Data Bank (PDB), CASP (Critical Assessment of protein Structure Prediction), Seoul National University, Google
- Publication Authors: John Jumper, et al.
- Technical background required: High
- Estimated read time (original text): 45 minutes
- Sentiment score: 85%, very positive (100% being most positive)
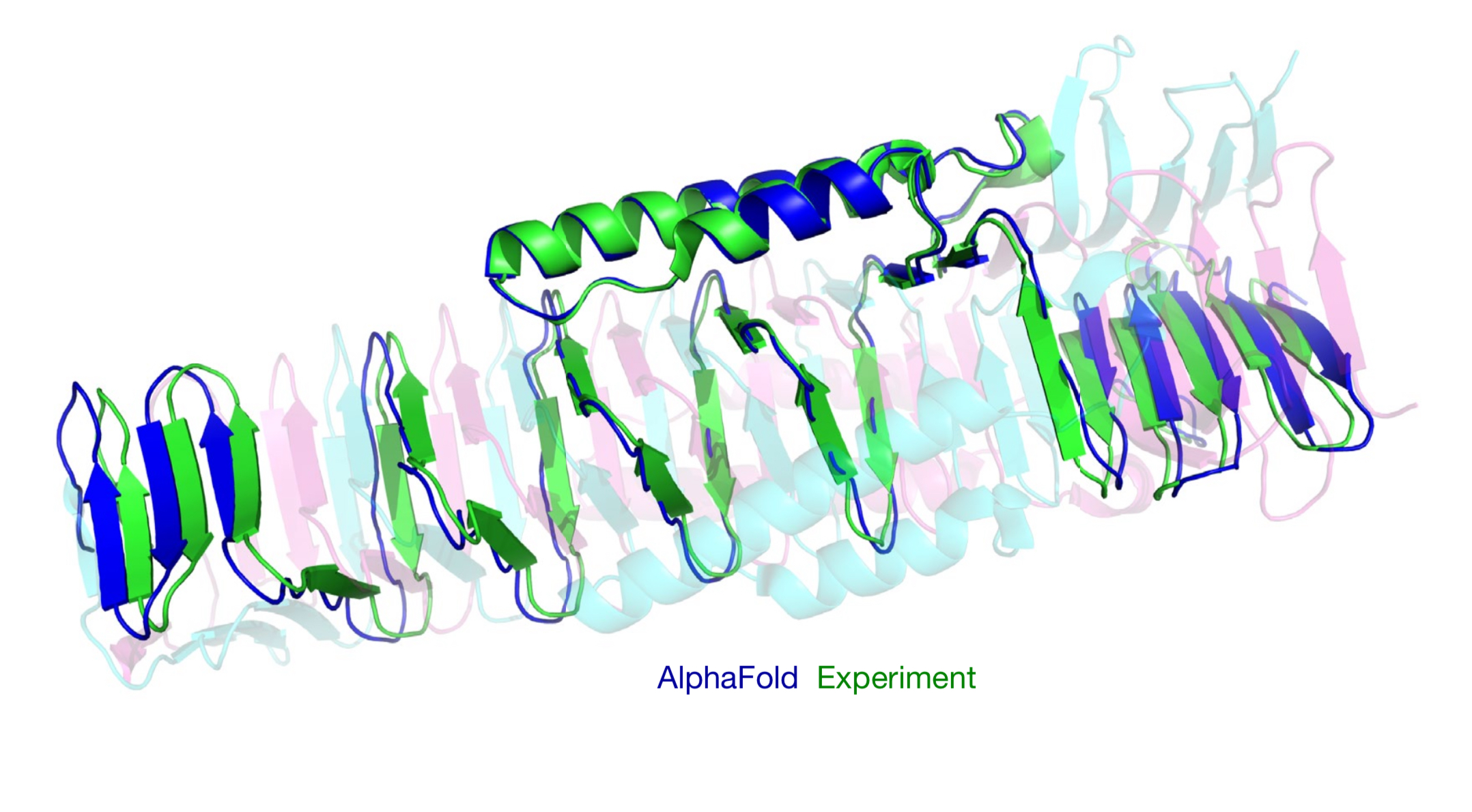
AlphaFold, developed by DeepMind, is an AI system that predicts three-dimensional protein structures from amino acid sequences. This tool addresses a 50-year-old challenge in biology, aiming to accelerate the traditionally slow and expensive process of protein structure determination. Utilizing deep learning and extensive genomic databases, AlphaFold provides accurate structural predictions within days.
Its performance is remarkable, achieving a median backbone accuracy of 0.96 Å RMSD (Root Mean Square Deviation, a measure of the average distance between atoms of superimposed proteins), compared to the next best method’s 2.8 Å. The system also has demonstrated high accuracy for 98.5% of human proteins and has been applied to over 500,000 proteins, including entire proteomes. Designed for researchers in structural biology, biochemistry, and pharmaceutical development, its potential applications include speeding up drug discovery, enhancing understanding of disease mechanisms, and advancing synthetic biology.
Goal of the Study: The study aimed to achieve near-experimental accuracy (below 1 Å RMSD) using a novel deep learning architecture that integrates evolutionary information, physical constraints, and geometric reasoning. This system would include an end-to-end trainable system that can handle diverse protein types, including those with limited evolutionary data, and predict structures for proteins resistant to traditional methods. Furthermore, the research sought to demonstrate AlphaFold’s ability to generate high-confidence predictions for entire proteomes efficiently.
Methodology:
1) Evoformer Architecture:
- Objective: To create a neural network module capable of processing evolutionary information from multiple sequence alignments (MSAs) and pairwise features to build a rich representation of protein structure.
- Method: The researchers developed the Evoformer, a novel neural network block that iteratively refines representations of the input data. It consists of:
- MSA stack: Processes evolutionary information from aligned sequences
- Pair stack: Handles pairwise relationships between residues
- Communication between stacks: Allows information to flow between the MSA and pair representations. The Evoformer uses attention mechanisms, including axial attention and triangle multiplication, to capture complex relationships in the data. It’s applied 48 times in sequence to progressively refine the protein structure prediction.
- Connection to Goal of the Study: The Evoformer addresses the challenge of integrating evolutionary and pairwise information effectively, which is crucial for accurate protein structure prediction. Its iterative refinement approach allows the model to capture subtle patterns in the data, contributing to the high accuracy of AlphaFold’s predictions.
- Analogy: Think of the Evoformer as a group of detectives working on a complex case. The MSA stack is like analyzing family histories, while the pair stack examines relationships between individuals. The communication between stacks is like detectives sharing clues. As they go through multiple rounds of investigation (48 iterations), they build a more complete and accurate picture of the situation (protein structure).
2) Structure Module:
- Objective: To transform the abstract representations from the Evoformer into explicit 3D protein structures.
- Method: The Structure Module operates on a “residue gas” representation, where each amino acid is treated as an independent rigid body. It uses:
- Invariant Point Attention (IPA): A geometry-aware attention mechanism that updates residue representations based on their 3D positionsFrame updates: Computes updates to the rotation and translation of each residue’s coordinate frame
- Side chain predictions: Predicts the angles of amino acid side chains. The module is applied iteratively (8 times) to refine the 3D structure prediction.
- Connection to Goal of the Study: The Structure Module enables AlphaFold to generate accurate 3D coordinates for protein structures, directly addressing the core goal of the study. Its ability to reason about geometric relationships in 3D space is key to achieving near-experimental accuracy.
- Analogy: Imagine assembling a complex 3D puzzle. The Structure Module is like a master puzzle-solver who can look at the pieces (residues) from different angles, adjust their positions (frame updates), and even reshape them slightly (side chain predictions) to make them fit better. By repeatedly examining and adjusting the puzzle (8 iterations), it creates a highly accurate final structure.
3) End-to-End Training:
- Objective: To create a unified system that can learn to predict protein structures directly from sequence data, without relying on hand-crafted features or intermediate steps.
- Method: The researchers designed a loss function called Frame-Aligned Point Error (FAPE) that compares predicted atom positions to true positions under many different alignments. They also incorporated auxiliary losses for:
- Predicted Local Distance Difference Test (pLDDT): Estimates per-residue model confidence
- Predicted TM-score (pTM): Estimates the global model quality
- Masked MSA prediction: Helps the model learn sequence patterns
The entire network is trained end-to-end on a dataset of known protein structures from the Protein Data Bank (PDB).
- Connection to Goal of the Study: End-to-end training allows AlphaFold to learn complex relationships between sequence and structure directly from data, contributing to its high accuracy and generalization ability. The carefully designed loss functions ensure that the model optimizes for biologically relevant structural features.
- Analogy: Think of end-to-end training as teaching a chef to create a complex dish. Instead of teaching separate lessons on ingredient selection, cutting techniques, and cooking methods, you let the chef practice making the entire dish from start to finish. The FAPE loss is like a discerning food critic who judges the dish from multiple perspectives, while the auxiliary losses are like additional judges focusing on specific aspects of the meal. This comprehensive training approach results in a chef (or AI) that can produce excellent results consistently.
4) Self-Distillation and Unlabeled Data Usage:
- Objective: To improve the model’s performance by leveraging a large amount of unlabeled protein sequence data.
- Method: The researchers employed a self-distillation approach:
- Train an initial model on PDB structures
- Use this model to predict structures for ~350,000 diverse sequences from Uniclust30
- Filter these predictions to a high-confidence subset
- Train a new model from scratch using both PDB data and the predicted structures.
- They also incorporated a BERT-style objective to predict masked elements of the MSA sequences during training.
- Connection to Goal of the Study: This approach allows AlphaFold to learn from a much larger set of proteins than just those with experimentally determined structures, improving its ability to handle diverse protein types and potentially increasing its accuracy on hard targets.
- Analogy: Imagine a student learning a new language. Initially, they learn from a textbook (PDB structures), but then they start watching movies in that language with subtitles (predicted structures). Even though the subtitles might not be perfect, they still help the student learn. The BERT-style objective is like covering up words in the subtitles and asking the student to guess them, further enhancing their understanding.
Key Findings & Results:
- Unprecedented Accuracy: AlphaFold achieved a median backbone accuracy of 0.96 Å RMSD on the CASP14 dataset, significantly outperforming the next best method (2.8 Å RMSD). This near-experimental level of accuracy represents a major advancement in protein structure prediction.
- Generalization to Diverse Proteins: The system demonstrated high accuracy across a wide range of protein types, including those with limited evolutionary information. It successfully predicted structures for 98.5% of the human proteome with high confidence.
- Rapid Prediction: AlphaFold can generate predictions in days, compared to the months or years often required for experimental structure determination. For a typical protein of 384 residues, it takes about 10 minutes on a GPU.
- Reliable Confidence Estimates: The pLDDT and pTM scores provided by AlphaFold correlate strongly with actual prediction accuracy, allowing researchers to assess the reliability of predictions.
- Effective Use of Evolutionary Information: The system showed a clear relationship between prediction accuracy and the depth of available multiple sequence alignments, with a notable improvement when the median alignment depth exceeds about 30 sequences.
These results demonstrate that AlphaFold has achieved a major milestone in protein structure prediction, offering a tool that could significantly accelerate research across various fields of biology and medicine.
Thinking Critically
Applications:
- Drug Discovery: AlphaFold could accelerate drug development by quickly predicting structures of potential drug targets, enabling faster identification of promising compounds and reducing time and costs in pharmaceutical research.
- Disease Research: Rapid protein structure prediction could enhance understanding of diseases at the molecular level, potentially revealing new therapeutic approaches for conditions like Alzheimer’s and cancer.
- Protein Engineering: In synthetic biology, AlphaFold could guide the design of novel proteins with specific functions, aiding in the development of new enzymes for industrial processes or proteins for environmental applications.
- Structural Biology: AlphaFold predictions could complement experimental methods, serving as starting models for X-ray crystallography or cryo-electron microscopy studies, particularly useful for challenging proteins like membrane proteins.
- Agricultural Biotechnology: The tool could assist in developing crops with improved traits by predicting plant protein structures, potentially leading to increased crop yield, nutritional value, or stress tolerance.
Broader Implications:
- Democratization of Research: AlphaFold’s accessibility could level the playing field in biotechnology research, allowing smaller labs without expensive equipment to contribute significantly to the field.
- Interdisciplinary Integration: The success of AlphaFold may accelerate the integration of AI and computational methods in scientific research, potentially shifting methodologies across various disciplines.
- Ethical Considerations: As AI becomes more powerful in genomics and proteomics, new discussions may arise regarding data ownership, privacy, and the ethical use of these technologies in biotechnology and medicine.
Limitations & Future Research Directions:
- Protein Complexes: AlphaFold currently has limitations in predicting structures of multi-protein complexes or protein interactions with other molecules. Future research could focus on extending its capabilities to these more complex systems.
- Dynamic Structures: The model predicts static structures, but many proteins are dynamic. Future work could aim to capture this flexibility, possibly by integrating molecular dynamics simulations or developing new AI approaches for multiple structural states.
- Interpretability: While highly accurate, understanding how AlphaFold arrives at its predictions remains challenging. Improving the model’s interpretability could provide insights into fundamental principles of protein folding and contribute to our understanding of protein biophysics.
Glossary
- Evoformer: A novel neural network block that iteratively refines representations of protein sequence and structural data through multiple attention mechanisms.
- Invariant Point Attention (IPA): A geometry-aware attention mechanism that updates residue representations based on their 3D positions while maintaining invariance to global rotations and translations.
- Frame-Aligned Point Error (FAPE): A loss function that compares predicted atom positions to true positions under many different alignments, encouraging accurate local structure prediction.
- Residue gas: A representation where each amino acid is treated as an independent rigid body, allowing for flexible modeling of protein structure.
- pLDDT (predicted Local Distance Difference Test): A per-residue confidence score that estimates the accuracy of the local structure prediction.
- pTM (predicted TM-score): A global confidence score that estimates the overall quality of the predicted protein structure.
- AlphaFold: The name of the AI system developed by DeepMind to predict protein structures from amino acid sequences.
No hype. No doom. Just actionable resources and strategies to accelerate your success in the age of AI.
 Join hosts Anthony, Shane, and Francesca for essential insights on AI's impact on jobs, careers, and business. Stay ahead of the curve – listen now!
Join hosts Anthony, Shane, and Francesca for essential insights on AI's impact on jobs, careers, and business. Stay ahead of the curve – listen now!
Recommended Research Reports


No hype. No doom. Just actionable resources and strategies to accelerate your success in the age of AI.
Join hosts Anthony, Shane, and Francesca for essential insights on AI's impact on jobs, careers, and business. Stay ahead of the curve – listen now!
Outsider
Labs.
A management consulting team focused on AI transformations for executives and business owners.
Work with us →